AMP PD Harmonized Data
Image
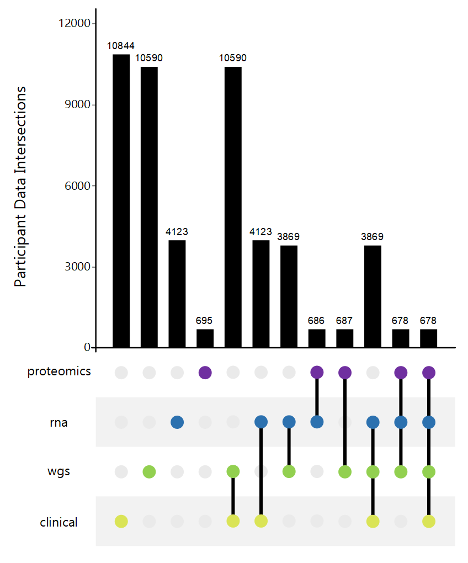
AMP PD generates and consolidates data from eight unified cohorts (BioFIND, HBS, LBD, LCC, PDBP, PPMI, STEADY-PD3 and SURE-PD3). Data was generated using standardized technology and centrally harmonized and quality controlled. All data was generated from samples collected under similar protocols.
This data harmonization process and single data use policy facilitates and simplifies cross-cohort analysis.
AMP Harmonized Data includes, but is not limited to:
- Clinical Data
- Demographic
- Enrollment
- Medical History
- MDS-UDPRS
- MoCA
- UPSIT
- Transcriptomic Data
- RNASeq from whole blood
- Illumina Novaseq sequencing
- Gencode v29 reference
- Genomic Data
- Whole Genome Sequencing from whole blood
- Illumina XTen sequencing
- Human Genome hg38 reference
- Proteomic Data
- Targeted proteomics from CSF and plasma
- Olink Explore analysis
- Untargeted proteomics
- Single Nucleus Brain Data
- Clinical Data
- Whole Genome Sequencing
- Single Nucleus RNA Sequencing